
ATROFIA MUSCULAR ESPINHAL
É uma doença degenerativa de origem genética. É uma das mais frequentes do sistema nervoso central e a mais comum das doenças autossómicas recessivas. Estima-se que esta atrofia afete 1 indivíduo em cada 10000 nascimentos. Esta doença é autossómica recessiva, logo, para que o descendente seja portador da doença é necessário que ambos os progenitores portem o gene responsável pela mutação.
Existem quatro tipos diferentes de AME:
-
Tipo I, AME infantil ou doença de Werdnig-Hoffmann
-
Tipo II, ou AME intermediaria
-
Tipo III, AME juvenil ou doença de Kugelbert-Welander
-
Tipo IV ou AME adulta.
O primeiro tipo pode manifestar-se desde a fase intra-uterina até aos 3 meses de vida extra-uterina. Caracteriza-se por ser o tipo mais grave.
Durante a gravidez não se verifica muita movimentação do feto, e o recém-nascido manifesta fraqueza acentuada nas musculaturas proximal e intercostal. Normalmente, os pacientes não ultrapassam os três anos de idade, e a principal causa de morte é o comprometimento do desenvolvimento do sistema respiratório. As crianças têm também dificuldade em se sentar sem apoio, apresentado um afundamento do osso esterno Outros sintomas incluem: dificuldade de deglutição e sucção; pernas mais fracas do que os braços; aumento da suscetibilidade a infeções respiratórias; acúmulo de secreções nos pulmões e garganta.
No tipo II os sintomas aparecem entre os 3 e os 15 meses de vida. A criança adquire a habilidade de sentar, desde que seja colocada nessa posição, mas não chega a conseguir andar, na maioria dos casos.
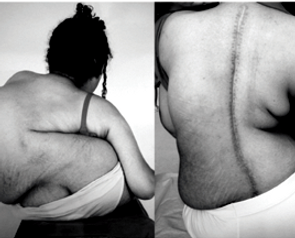
No tipo III, os sintomas surgem entre os dois e os 17 anos de idade, e é caracterizada pelo comprometimento dos membros superiores. A sua evolução é lenta, no entanto os portadores, normalmente, necessitam de usar instrumentos que auxiliem a sua locomoção, como muletas e até cadeiras de rodas.
O tipo IV afeta adultos entre os 30 e os 40 anos, e é o menos grave. A apresentação dos sintomas acontece de forma lenta e leva ao total comprometimento muscular.

Diagnóstico
O diagnóstico para esta doença é feito por meio de três exames:
Eletromiografia, exame que mede a atividade elétrica do músculo;
Biópsia muscular, na qual se observa degeneração da fibra muscular em diferentes estágios;
Análise do cromossoma cinco, que procura a deleção do gene SMN. Quando a realização deste exame é possível (análise do cromossoma cinco) torna-se desnecessário realizar os outros dois anteriores.

Causas
A AME está relacionada com o gene SMN (survival of motor neuron) que está localizado no cromossoma cinco. Este gene tem nove exões que codificam 254 proteínas. Uma cópia está na região centromérica e outra na região telomérica. O gene recebe o nome de SMN1 e a sua cópia de SMN2.
A deleção do gene SMN1 é o que define a apresentação dos sintomas, e a quantidade de gene SMN2 é o que determina a gravidade dos sintomas que irão aparecer.
Tratamento
Nas formas graves (tipo 1) o tratamento é baseado em oferecer suporte clínico à criança e emocional à família. Nos tipos II, III, IV os graus de envolvimento são variáveis e as formas de tratamento são estabelecidas conforme a dificuldade de cada criança/adulto.